5. Day 4: Introduction to Automation and Nextflow¶
5.1. Overview¶
5.1.1. Lead Instructor¶
5.1.2. Co-Instructor(s)¶
5.1.4. General Topics¶
- Introduction to Nextflow
- Use of workflow systems for automation / reproducibility
- Basic syntax of Nextflow
- Transform and execute a workflow in Nextflow
5.2. Schedule¶
- 09:00 - 10:00 Introduction to Nextflow
- 10:00 - 11:00 Parameters, Channels and Processes
- 11:00 - 11:30 Coffee break
- 11:30 - 12:30 Docker/Singularity, Executors and Channel Operations
- 12:30 - 14:00 Lunch break
- 14:00 - 15:00 Practical exercises in Nextflow
- 15:00 - 16:00 Practical exercises in Nextflow
- 16:00 - 16:15 Coffee break
- 16:15 - 18:00 Practical exercises in Nextflow
5.3. Learning Objectives¶
- Find and use Nextflow tool definitions online.
- Understand how to write Nextflow scripts and definitions for command line tools.
- Understand the concepts of Nextflow
Channels,ProcessesandChanneloperators. - Understand how to handle multiple inputs and outputs in Nextflow.
- Understand Nextflow’s configuration file (
nextflow.config), profiles and input parameters. - Use Docker/Singularity with Nextflow to provide software dependencies and ensure reproducibility.
- Join Nextflow tools into a workflow.
- Run Nextflow workflows on local, HPC and cloud systems.
Nextflow: A Tutorial Through Examples
Reproducible workflows using Nextflow
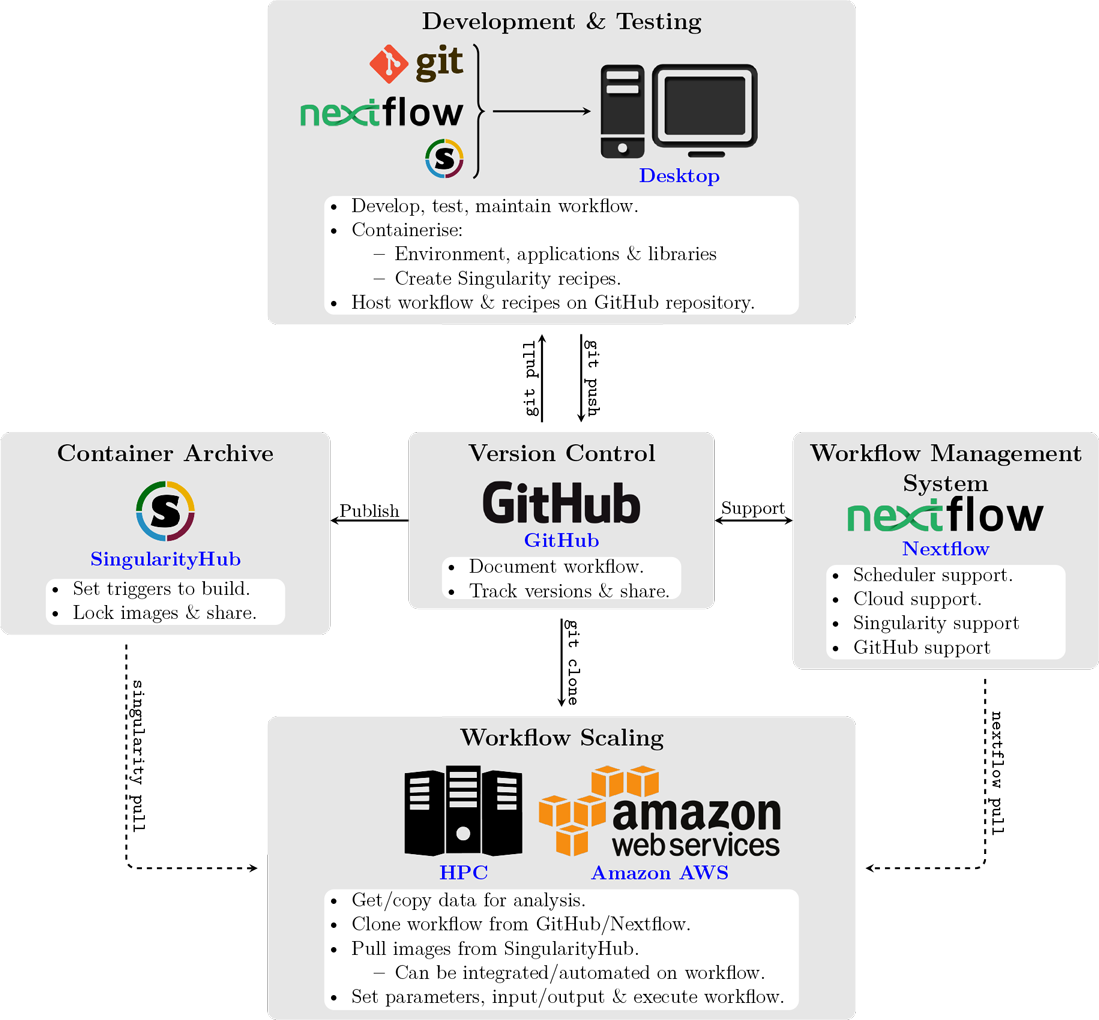 Flow chart summarizing the resources and best practices for development, maintenance, sharing and publishing of reproducible and portable workflows.
Flow chart summarizing the resources and best practices for development, maintenance, sharing and publishing of reproducible and portable workflows.
5.4. 1. Introduction¶
This tutorial in an introduction to Nextflow, primarily through examples. Since the tutorial is brief, it is designed to whet your appetite – we’re only going to dip in and out of some of its features in a superficial way.
Exercises: Throughout this tutorial there will be some practical examples. Not all will be covered in class for time reasons but you can come back and do them.
5.4.1. 1.1 Workflow Languages¶
Many scientific applications require:
- Multiple data files
- Multiple applications
- Perhaps different parameters
General purpose languages not well suited:
- Too low a level of abstraction
- Does not separate workflow from application
- Not reproducible
Nextflow is a Groovy-based language for expressing workflows:
- Portable – works on most Unix-like systems
- Very easy to install (NB: requires Java 7, 8)
- Scalable
- Supports Docker/Singularity
- Supports a range of scheduling systems
Key Nextflow concepts:
- Processes: actual work being done – usually simple (call program that does the analysis)
- Channels: for communication between processes (inputs, outputs)
- When all inputs ready, process is executed.
- Each process runs in its own directory – files are staged.
- Supports resumption of previous partial runs.
5.4.2. 1.2. Nexflow Script¶
First, lets set up a directory where we will do all our Nextflow exercises:
mkdir $HOME/day4
cd $HOME/day4
Then, we download the data we will be using for the exercises:
wget https://github.com/fpsom/CODATA-RDA-Advanced-Bioinformatics-2019/raw/master/files/data/tutorial.zip
unzip tutorial.zip
Tyep ls -l and hit <ENTER> to view the contents of your directory. Your day4 directory will now contain Nextflow scripts (ending with .nf) and a data folder that we will use in this tutorial.
day4
|--cleandups.nf
|--data
| |--11.bim .. 14.bim
| |--2016-REG-01.dat .. 2019-XTR-12.dat
| |--pop
| | |--BEB.[bed,bim,fam]
| | |--CEU.[bed,bim,fam]
| | |--JPT.[bed,bim,fam]
| | |--YRI.[bed,bim,fam]
|--solutions
| |--ex1-cleandups.nf
| |--ex2-cleandups.nf
| |--ex3-groovy.nf
| |--ex4-cleandups.nf
| |--ex5-weather.nf
| |--ex6-dockersee.nf
Exercise 1: You have an input file with 6 columns (see below), where column 2 is an “index” column. Identify rows that have identical indexes (column 2) and remove them from the file. Your input file looks like this:
11 11:189256 0 189256 A G
11 11:193788 0 193788 T C
11 11:194062 0 194062 T C
11 11:194228 0 194228 A G
11 11:193788 0 193788 A C
Let’s have a look at this file:
less -S data/11.bim
Solution - using bash:
cut -f 2 data/11.bim | sort | uniq -d > dups
grep -f dups data/11.bim > 11.clean
This is easy to do in bash - very simple example, not realistic for Nextflow
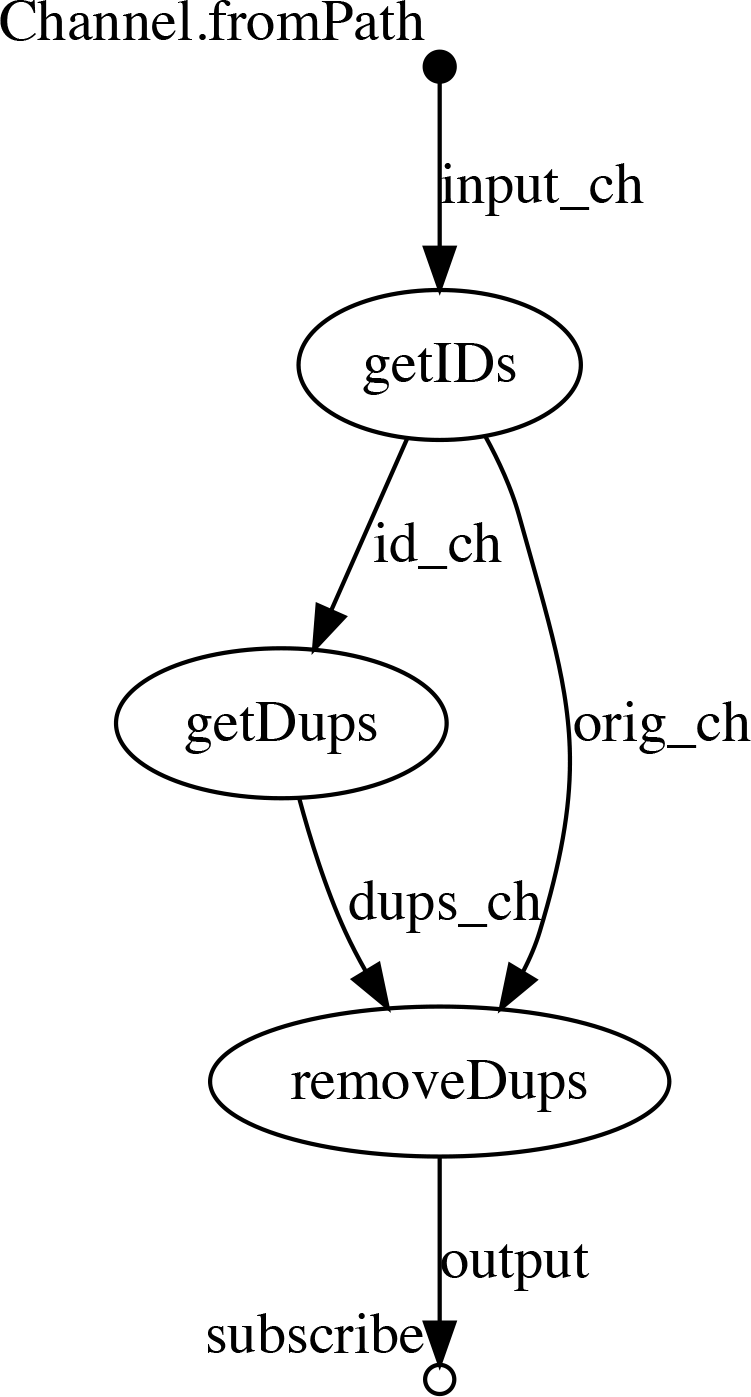
Solution - using Nextflow:
#!/usr/bin/env nexflow
input_ch = Channel.fromPath("data/11.bim")
process getIDs {
input:
file input from input_ch
output:
file "ids" into id_ch
file "11.bim" into orig_ch
script:
"cut -f 2 $input | sort > ids"
}
process getDups {
input:
file input from id_ch
output:
file "dups" into dups_ch
script:
"""
uniq -d $input > dups
touch ignore
"""
}
process removeDups {
input:
file badids from dups_ch
file orig from orig_ch
output:
file "clean.bim" into output
script:
"grep -v -f $badids $orig > clean.bim "
}
output.subscribe { print "Done!" }
Using a text editor like emacs or vim, open the Nextflow script cleandups.nf and have a look at it.
NB: The use of Nextflow variables – within a double quoted string, there is string interpolation marked with the $. If you want to access a system environment variable you need to also escape with a backslash. So in the Nextflow program, you can normally just refer to Nextflow variables unadorned with their names (e.g. $input) and environment variables with a dollar (e.g. $HOME) but within a double/triple-quoted string it’s \$input and \$HOME. File names can be relative (to the current working directory where the script is being run in, not to the location of the script), or absolute. Great care needs to be taken with using absolute path names since this reduces the portability of scripts, particualarly when you are using Docker.
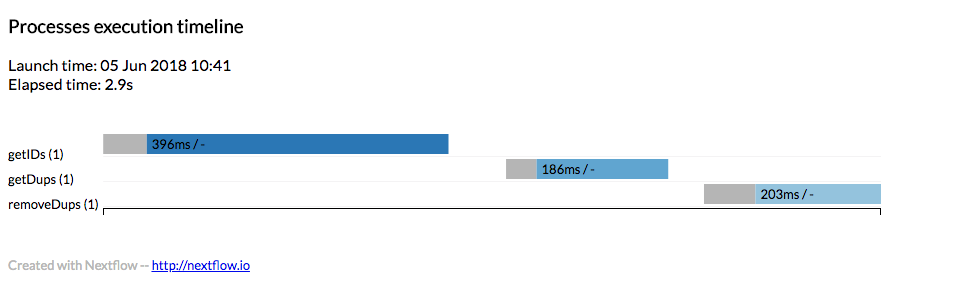
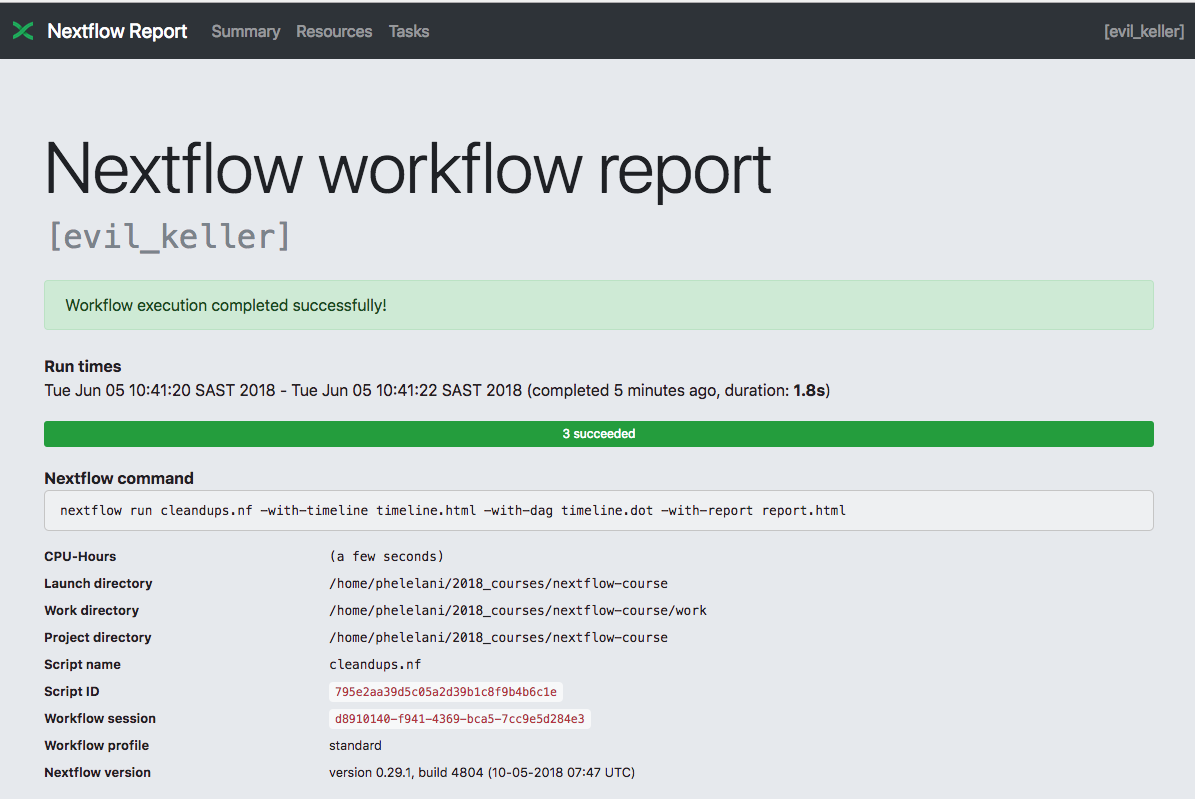
Now we can execute our script:
nextflow run cleandups.nf
The output we get:
N E X T F L O W ~ version 19.07.0
Launching `cleandups.nf` [distraught_lamarr] - revision: fb99ce6125
executor > local (3)
[b3/aa0380] process > getIDs (1) [100%] 1 of 1 ✔
[90/cebf36] process > getDups (1) [100%] 1 of 1 ✔
[9c/e0cb7d] process > removeDups (1) [100%] 1 of 1 ✔
Done!
NB: Nextflow creates a work directory, and inside of that are the working directories of each process – in the example above you can see that the getIDs process was launched in a directory with a prefix aa0380, inside the directory b3. The directory structure is looks like:
day4
|--cleandups.nf
|--data
| |--11.bim .. 14.bim
| |--2016-REG-01.dat .. 2019-XTR-12.dat
| |--pop
| | |--BEB.[bed,bim,fam]
| | |--CEU.[bed,bim,fam]
| | |--JPT.[bed,bim,fam]
| | |--YRI.[bed,bim,fam]
|--solutions
| |--ex1-cleandups.nf
| |--ex2-cleandups.nf
| |--ex3-groovy.nf
| |--ex4-cleandups.nf
| |--ex5-weather.nf
| |--ex6-dockersee.nf
|--work
| |--90
| | |--cebf3649d883f88381e32b4912b560
| | | |--ids -> /Users/phele/day4/work/b3/aa0380f2a1bca447259b7ffd390083/ids
| | | |--ignore
| |--9c
| | |--e0cb7d8d26682d7d4a1c44392f2bb3
| | | |--11.bim -> /Users/phele/day4/data/11.bim
| | | |--clean.bim
| | | |--dups -> /Users/phele/day4/work/90/cebf3649d883f88381e32b4912b560/dups
| |--b3
| | |--aa0380f2a1bca447259b7ffd390083
| | | |--11.bim -> /Users/phele/day4/data/11.bim
| | | |--ids
The names of the working directory are randomly chosen so if you run it, you will get different names. Also, each time you run a process ever, it will get a unique working directory. There is no danger of name clashes Instead of naming the file you get from a channel you can also:
- specify
stdinif your process expects data to come fromstdinrather than a named file. Nextflow will pipe the file to standard input; - specify
stdoutif your process produces data onstdoutand you want that data to go into thechannel
Exercise 2: Change the script so that you use stdin or stdout in the getIDs and getDups processes to avoid the use of the temporary file ids. You can see the solution here
5.4.3. 1.3. Partial Execution¶
If execution of workflow is only partial (e.g., because of error), only need to resume from process that failed:
nextflow run cleandups.nf -resume
5.5. 2. Groovy¶
Can inter-mix Nextflow, Groovy and Java code
- Very powerful, flexible
- Don’t need to know much (any?) Groovy but a little knowledge is a powerful thing Here we’ll do some cookbook Groovy…
5.5.1. 2.2. Groovy Closures¶
Closures are anonymous functions – similar to lambdas in Python.
- Don’t want the overhead of naming a function we only use once
- Typically use with higher-order functions – functions that take other functions as arguments. Very powerful and useful
Syntax for a closure that takes one argument:
{ parm -> expression }
This is an anonymous function that takes one parameter (I’ve called it parm, you can call it whatever you want) and expression is a valid expression, probably including the parameter. Let’s look at some examples:
{ a -> a*a } (3)
{ a -> a*a+7*a - 2 } (3)
for (n in 1..5) print( {it*it} (n));
{ x, y -> Math.sqrt(x*x + y*y) } (3,4)
OK, I am simplifying a bit here. Closures are a bit more than functions.
Now what we have seen so far isn’t so useful but the power comes when we have a function that take another function. Consider this very simplistic example. Suppose we have a program where we are manipulating lists of numbers. Sometimes we want to sum a list, sometimes we want to sum the squares of the numbers; sometimes we want to sum the cubes of the cosines of the numbers. The business of going through the list and summing is the same in all cases. What differs is what we do to the numbers in each case – so rather than have a separate procedure for each type of summation, we just have one. But we pass this procedure a function that says what to do:
int doX(f, nums) {
sum=0
for ( n in nums ) {
sum = sum+f(n)
}
return sum
}
We can call it thus:
print doX ( {a->a}, [4,5,16] );
print doX ( {a->a*a}, [4,5,16] );
print doX ( { it*it }, [4,5,16]);
m=10
print doX({a->m*a+2}, [1,2,3])
NB: You don’t have to name the parameter – if you don’t name the parameter then the name it is assumed.
Exercise 3: Look at the sample Groovy code here. Try to understand and execute on your machine.
5.6. 3. Generalising and Extending¶
We’ll now extend this example, introducing more powerful features of Nextflow as well as some of the complication of workflow design.
Extending the example:
- Parameterise the input.
- Want output to go to convenient place.
- Workflow takes in multiple input files – processes are executed on each in turn.
- Complication: may need to carry the base name of the input to the final output.
- Can repeat some steps for different parameters.
5.6.1. 3.1. Parameters¶
Parameters can be specified in a Nextflow script file:
input_ch = Channel.fromPath(params.data_dir)
They can also be passed to the Nextflow command when executing a script:
nextflow run phylo1.nf --data_dir data/polyseqs.fa
During debugging you may want to specify default values:
params.data_dir = 'data'
If you run the Nextflow program without parameters, it will use this as a default value; if you give it parameters, the default value is replaced. Of course, as a matter of good practice, default values for parameters are really designed for real parameters of the process (like gap penalties in a multiple sequence alignment) rather than data files.
Nextflow makes a distinction between parameters with a single dash (-) and with a double dash (--). The single dash ones are from a small, language defined subset modifying the behaviour of Nextflow – for example we’ve seen --with-dag already.
The double-dash parameters are user-defined and completely extensible – they are used to populate params. They modify the behaviour of your program.
5.6.2. 3.2. Channels¶
Nextflow channels support different data types:
filevalset
NB: val is the most generic – could be a file name. But sending a file provides power since you can access Groovy’s file handling capacity and, more importantly does staging of files
5.6.2.1. 3.2.1. Creating channels¶
Channel.create()
Channel.empty
Channel.from("blast","plink")
Channel.fromPath("data/*.fa")
Channel.fromFilePairs("data/{YRI,CEU,BEB}.*)
Channel.watchPath("*fa")
There are others.
NB: The fromPath method takes a Unix glob and creates a new channel which has all the files that match the glob. These files are then emitted one by one to processes that use these values. This default semantics can be changed using the channel operators that Nexflow provides, some of which are shown below. There are many, many operations you can do on channels and their contents.
bind buffer close
filter map/reduce group
join, merge mix copy
split spread fork
count min/max/sum print/view
5.6.3. 3.3. Generalising Our Example¶
5.6.3.1. 3.3.1. Multiple inputs¶
#!/usr/bin/env nextflow
params.data_dir = "data"
input_ch = Channel.fromPath("${params.data_dir}/*.bim")
process getIDs {
input:
file input from input_ch
output:
file "${input.baseName}.ids" into id_ch
file "$input" into orig_ch
script:
"cut -f 2 $input | sort > ${input.baseName}.ids"
}
process getDups {
input:
file input from id_ch
output:
file "${input.baseName}.dups" into dups_ch
script:
out = "${input.baseName}.dups"
"""
uniq -d $input > $out
touch ignore
"""
}
process removeDups {
publishDir "output", pattern: "${badids.baseName}_clean.bim", overwrite:true, mode:'copy'
input:
file badids from dups_ch
file orig from orig_ch
output:
file "${badids.baseName}_clean.bim" into cleaned_ch
script:
"grep -v -f $badids $orig > ${badids.baseName}_clean.bim "
}
Here the getIDs process will execute once, for each file found in the initial glob. On a machine with multiple cores, these would probably execute in parallel, and as we’ll see later if you are running on the head node of a cluster, each could run as a separate job.
NB: that in this version of getIDs we name the output file dependant on the input file. This is convenient to do because now we are taking many input files. There is no danger of there being any name clashes during execution because each parallel execution of getIDs runs in a separate local directory. However, at the end we want to be able to distinguish which output came from which input without having to do detective work – so we name the files conveniently. Files that get created on the way but don’t need at the end we can name boringly.
nextflow run cleandups.nf
N E X T F L O W ~ version 19.07.0
Launching `cleanups.nf` [small_wozniak] - revision: f8696171b0
executor > local (3)
[6c/1b5ca2] process > getIDs (1) [100%] 1 of 1 ✔
[74/7d0dc8] process > getDups (1) [100%] 1 of 1 ✔
[05/51ca59] process > removeDups (1) [100%] 1 of 1 ✔
Now I’m going to add a next step – say we want to split the IDs into groups using split but try different values of splitting.
5.6.3.2. 3.3.2. Multiple parameters¶
Exercise 4: Now try adding a process to our Nextflow example and for splitting the file but using different split values (SOLUTION HERE), e.g.:
split -l 400 data.txt dataX
will produce files dataXaa, dataXab, dataXac and so on…
splits = [400,500,600]
process splitIDs {
input:
file bim from cleaned_ch
each split from splits
output:
file ("*-$split-*") into output_ch;
script:
"split -l $split $bim ${bim.baseName}-$split- "
}
Have a look at the modified Nextflow scrip here.
5.6.4. 3.4. Managing Grouped Files¶
We’ve seen so far where we have a stream of file being processed independently. But in many applications there may be matched data sets. We’ll now look at an example, using a popular bioinformatics tool called PLINK. In its most common usages, PLINK takes in three related files, typically with the same but different suffixed: .bed, .bim, .fam.
Short version of the command:
plink --bfile /path/YRI --freq --out /tmp/YRI
Long version of the command:
plink --bed YRI.bed --bim YRI.bim --fam YRI.fam --freq --out /tmp/YRI
If you don’t know what PLINK does, don’t worry. It’s the Swiss Army knife for bioinformatics. The above commands are equivalent (the first is the short-hand for the second when the bed, bim, and fam files have the same base). The command finds frequencies of genome variations – the output in this example will go into a file called YRI.frq.
Problem:
- Pass the files on another channel(s) to be staged
- Pass the base name as value/or work it out
Pros/Cons
- Simple
- Need extra channel/some gymnastics
Lets recap – Groovy closures. Simply, a closure is an anonymous function:
- Code wrapped in braces
{ } - Default argument called
it
[1,2,3].each { print it * it }
[1,2,3].each { num -> print num * num }
Similar to lambdas in Python and Java.
5.6.4.1. 3.4.1. Version 1: map¶
#!/usr/bin/env nextflow
params.dir = "data/pops/"
dir = params.dir
params.pops = ["YRI","CEU","BEB"]
Channel.from(params.pops)
.map { pop ->
[ file("$dir/${pop}.bed"), file("$dir/${pop}.bim"), file("$dir/${pop}.fam")]
}
.set { plink_data }
plink_data.subscribe { println "$it" }
This example takes a stream of values from params.pops and for each value (that’s what map does) it applies a closure that takes a string and produces a tuple of files. That tuple is then bound to a channel called plink_data. NB: There are two distinct uses of the set:
- As a channel operator as shown here
- In an input/output clause of a channel
[data/pops/YRI.bed, data/pops/YRI.bim, data/pops/YRI.fam]
[data/pops/CEU.bed, data/pops/CEU.bim, data/pops/CEU.fam]
[data/pops/BEB.bed, data/pops/BEB.bim, data/pops/BEB.fam]
Now let’s look at more realistic exammple. To try this example on your own computer.
NB: Since you may not have plink on your computer, our code actually fakes the output. If you do have plink you can make the necessary changes.
process getFreq {
input:
set file(bed), file(bim), file(fam) from plink_data
output:
file "${bed.baseName}.frq" into result
"""
plink --bed $bed \
--bim $bim \
--fam $fam \
--freq \
--out ${bed.baseName}"
"""
}
Look at plink1B.nf. It’s a slightly different ways of doing things. On examples of this size, none of these options are much better or worse but it’s useful to see different ways of doing things for later.
5.6.4.2. 3.4.2. Version 2: fromFilePairs¶
Use fromFilePairs
- Takes a closure used to gather
filestogether with the samekey:
x_ch = Channel.fromFilePairs( files ) { closure }
Specify the files as a glob. Closure associates each file with a key. fromPairs puts all files with same key together; returns a list of pairs (key, list)
#!/usr/bin/env nextflow
commands = Channel.fromFilePairs("/usr/bin/*", size:-1) { it.baseName[0] }
commands.subscribe { k = it[0];
n = it[1].size();
println "There are $n files starting with $k";
}
Here we use standard globbing to find all the files in the /usr/bin directory. The closure takes the first letter of each file – all the files with the same letter are put together. The size parameter says how many we put togther : -1 means all.
A more complex example – default closure
Channel
.fromFilePairs ("${params.dir}/*.{bed,fam,bim}", size:3, flat : true)
.ifEmpty { error "No matching plink files" }
.set { plink_data }
plink_data.subscribe { println "$it" }
fromFilePairs
- Matches the
glob - The first
*is taken as the matching key - For each unique match of the
*we return- The thing that matches the
* - The list of files that match the glob with that item
- Up to 3 matching files (the default of size is 2 – hence the name)
- The thing that matches the
[CEU, [data/pops/CEU.bed, data/pops/CEU.bim, data/pops/CEU.fam]]
[YRI, [data/pops/YRI.bed, data/pops/YRI.bim, data/pops/YRI.fam]]
[BEB, [data/pops/BEB.bed, data/pops/BEB.bim, data/pops/BEB.fam]]
process checkData {
input:
set pop, file(pl_files) from plink_data
output:
file "${pl_files[0]}.frq" into result
script:
base = pl_files[0].baseName
"plink --bfile $base --freq --out ${base}"
}
OR
process checkData {
input:
set pop, file(pl_files) from plink_data
output:
file "${pop}.frq" into result
script:
"plink --bfile $pop --freq --out $pop"
}
5.6.4.3. 3.4.3 Version 3: Final version¶
#!/usr/bin/env nextflow
params.dir = "data/pops/"
dir = params.dir
params.pops = ["YRI","CEU","BEB"]
Channel
.fromFilePairs("${params.dir}/{YRI,BEB,CEU}.{bed,bim,fam}",size:3) {
file -> file.baseName
}
.filter { key, files -> key in params.pops }
.set { plink_data }
process checkData {
input:
set pop, file(pl_files) from plink_data
output:
file "${pop}.frq" into result
script:
"plink --bfile $pop --freq --out $pop"
}
Exercise 5: Have a look at weather.nf. In the data directory are set of data files for different years and months. First, I want you to use paste to combine all the files for the same year and month (paste joins files horizontal-wise). Then these new files should be concated.
5.6.5. 3.5. On absolute paths¶
Great care needs to be taken when referring to absolute paths. Consider the following script. Assumming that local execution is being done, this should work.
input = Channel.fromPath("/data/batch1/myfile.fa")
process show {
input:
file data from input
output:
file 'see.out'
script:
cp $data /home/scott/answer
However, there is a big difference in the two uses of absolute paths. While it might be more appropriate or useful to pass the first path as a parameter, there is no real problem. Netflow will transparently stage the input files to the working directories as appropriate (generally using hard links). But the second hard-coded file will cause failures when we try to use Docker.
5.7. 4. Nextflow + Docker & Singularity Containers¶
Light-weight virtualisation abstraction layer:
- Currently run on
Unixlike systems (e.g., Linux, macOS). - Windows support coming…
You create Docker/Singularity images locally, or get from repositories (): Getting Docker images from repositories:
docker pull ubuntu
docker pull quay.io/banshee1221/h3agwas-plink
Getting Singularity images from repositories:
singularity pull docker://ubuntu
singularity pull docker://quay.io/banshee1221/h3agwas-plink
Running Docker
docker run <some-image-name>
Running Singularity
singularity exec <some-image-name>
Docker and Singularity often run images in background (e.g. webserver), but can also run interactively:
## Running Docker interactively
sudo docker run -t -i quay.io/banshee1221/h3agwas-plink
## Running Singularity interactively
singularity shell docker://quay.io/banshee1221/h3agwas-plink
Nextflow supports Docker & Singularity
- Well designed script should be highly portable
- Each process gets run as a separate Docker call (e.g, under the hood, a
docker runis called) - Can use the same or different Docker images for each process, parameterisable
Simple example (assuming all processes use the same Docker/Singularity image)
## For Docker
nextflow run plink2.nf -with-docker quay.io/banshee1221/h3agwas-plink
## For Singularity
nextflow run plink.nf -with-singularity docker://quay.io/banshee1221/h3agwas-plink
Now, even if you don’t have plink, your script will work because my Docker/Singularity image has plink insalled!
5.7.1. 4.1. Directory/file access¶
Nextflow Docker/Singularity support highly transparent – but pay attention to good practice:
- For each process Docker/Singularity mounts the work directory for that process on the Docker/Singularuty image.
- Files can be staged in and out using Nextflow mechanisms.
- Other files available: directories mounted through run time options or on the image
- No other files on the host machine including the current directory
- Process executes in the Docker/Singularity environment
#!/usr/bin/env nextflow
data = Channel.fromPath("data/pops/YRI.bim")
process see {
echo true
input:
file bim from data
output:
file count
publishDir params.publish, overwrite:true, mode:'move'
"""
hostname
echo "Path is \$( pwd )\n "
echo "Parent directory has \$( ls .. )\n"
echo "My home directory has \$( ls /home/phele )\n"
wc -l $bim > count
ls
"""
}
The output that is produced is:
N E X T F L O W ~ version 0.21.2
Launching show_env.nf
[warm up] executor > local
[94/597f09] Submitted process > see (1)
89ad448ae0b2
Path is /home/scott/witsGWAS/dockerized/work/94/597f09ca6cc01c7be015052f7f072c
Parent directory has 597f09ca6cc01c7be015052f7f072c
My home directory has witsGWAS
YRI.bim
count
NB: Although the script’s pwd shows /home/scott/witsGWAS/dockerized/work/94/597f09ca6cc01c7be015052f7f072c
- Only these specific directories are mounted
- Only the files in the innermost directory are available
Any absolute paths (other than those used in staging) will result in error.
A little more technical detail: In the above example, the YRI.bim file is staged into the working directory on the Docker image. To achieve this, the directories data and data/pop are mounted on the Docker image. But no other sub-directories of data are available. But all the other files in data/pops are available!.
Exercise 6: Have a look at dockersee.nf and run it like this:
nextflow run dockersee.nf -with-docker quay.io/banshee1221/h3agwas-plink
5.7.2. 4.2. Docker/Singularity profiles¶
In nextflow.config:
profiles {
...
docker {
docker.enabled = true
process.container = 'quay.io/banshee1221/h3agwas-plink:latest'
}
singularity {
singularity.enable = true
process.container =
}
}
For Docker, we can run:
nextflow run gwas.nf -profile docker
For Singularity, we can run:
nextflow run gwas.nf -profile singularity
Profiles can be extended in many ways:
- Different processes can use different containers
- Can mount other host directories
- Can pass arbitrary Docker parameters
5.8. 5. Executors¶
A Nextflow executor is the mechanism which Nexflow runs the code in each of the processes:
- Default is
local: process is run as a script
There are many others:
- PBS/Torque
- SLURM
- Amazon (AWS Batch)
- SGE (Sun Grid Engine)
5.8.1. 5.1. Selecting an executor¶
Can be done through annotating each process:
executordirective, e.g.executor 'pbs'- resource constraints.
Or, nextflow.config file:
- either global or per-process.
5.8.2. 5.2. Nextflow on a cluster (HPC)¶
Script runs on the head node!
- Nextflow uses the
executorinformation to decide how the job should run. - Each process can be handled differently
- Nextflow submits each process to the job scheduler on your behalf (e.g, if using
PBS/Torque,qsubis done)
What’s great about this is that Nextflow handles any dependencies – if some jobs rely on other jobs completing first, Nextflow handles this. There are also simple directives you can use to restrict the number of jobs running at a time so this can help you if there are queue restrictions. It’s possible to do this without Nextflow, but it’s much more transparent.
Example:
process {
executor = 'pbs'
queue = 'batch'
scratch = true
cpus = 5
memory = '2GB'
}
5.8.3. 5.3. Amazon Elastic Compute Cloud (EC2)¶
Netflow has native support for EC2. You need an account on EC2, and an image with the appropriate support.
To launch your code:
nextflow cloud create GenomeCloud -c 5
You need to put some information in your nextflow.config file, e.g., the Amazon Machine Image (AMI), your credentials, etc. In this example GenomeCloud is a name I have given — I am creating a cluster with one master and four worker nodes.
If successful, Nextflow will give you the name of the headnode of your cluster.
sshinto into it- run
nextflowon it.
Afterwards shut down:
nextflow shutdown GenomeCloud
5.9. 6. Channel Operations¶
Nextflow tries to maximise concurrency
- processes are by default synchronised by channels
- when data arrives on all input channels, process executes
No problem if channels only ever have one value – but when multiple values, may be an issue.
#!/usr/bin/env nextflow
Channel.fromPath("data/*.dat").set { data }
process P1 {
input:
file(data)
output:
file "${fbase}.pre" into channelA
file data into channelB
script:
fbase=data.baseName
"echo dummy > ${fbase}.pre"
}
process P2 {
input:
file pre from channelA
output:
file pre into channelC
script:
if (pre.baseName == "a")
"sleep 4"
else
"sleep 1"
}
process P3 {
echo true
input:
file(data) from channelB
file(pre) from channelC
script:
"""
echo "${data} - $pre"
"""
}
Solution: join/merge channels
x.merge(y): Items emmited by the channelsxandyare combined into a new channel.x.join(y): Items emmited by the channelsxandyare joined together into one channel based on existing matching key. Default: first element in each item.
5.9.1. 6.1. Using join:¶
ch1 = Channel.from( "a","b","c" )
ch2 = Channel.from( "a","d","e","a","c","b" )
ch1.join(ch2).subscribe { println it }
Yields:
a
b
c
5.9.1.1. 6.1.1 Using join - tuples:¶
ch1 = Channel.from( ["a",1], ["b",4], ["c",5] )
ch2 = Channel.from( ["a",10], ["d",8], ["e",7], ["a",9], ["c",1], ["b",10] )
ch1.merge(ch2).subscribe { println it }
Yields:
[a, 1, 10]
[b, 4, 10]
[c, 5, 1]
5.9.2. 6.2. Using merge¶
ch1 = Channel.from( "a","b","c" )
ch2 = Channel.from( "a","d","e","a","c","b" )
ch1.merge(ch2).subscribe { println it }
Yields:
[a, a]
[b, d]
[c, e]
5.9.2.1. 6.2.1. Using merge - tuples:¶
ch1 = Channel.from( ["a",1], ["b",4], ["c",5] )
ch2 = Channel.from( ["a",10], ["d",8], ["e",7], ["a",9], ["c",1], ["b",10] )
ch1.merge(ch2).subscribe { println it }
Yields:
[a, 1, a, 10]
[b, 4, d, 8]
[c, 5, e, 7]
5.9.3. 6.3. join vs. merge¶
join:
- If values are singletons, then the values must be the same
- If value is tuple if the, then the first element of the tuple must be the same
merge:
- Merges everything into a channel, no matching.
5.9.4. 6.4. Using join on our example:¶
process P3 {
echo true
input:
set file(pre), file(data) from channelB.join(channelC)
script:
"""
echo "${data} - $pre"
"""
}
This will not work. ChannelA, ChannelB and ChannelC all emmit single file. The join command will not work since files have different extension (even though the file.baseName are the same. We need to change our approach to this – Use tuples/list and set a baseName as an identifier for each file pair .pre and .dat.
5.9.4.1. 6.4.1. Working version of the example¶
#!/usr/bin/env nextflow
Channel.fromPath("data/*.dat").set { data }
process P1 {
echo true
input:
file(data)
output:
set val(data.baseName), file("${fbase}.pre") into channelA
set val(data.baseName), file(data) into channelB
script:
fbase=data.baseName
"echo dummy > ${fbase}.pre"
}
process P2 {
echo true
input:
set name, file(pre) from channelA
output:
set name, file(pre) into channelC
script:
if (pre.baseName = /.*TMP.*/)
"sleep 4"
else
"sleep 1"
}
process P3 {
echo true
input:
set name, file(data), file(pre) from channelB.join(channelC)
script:
"""
echo "${data} - ${pre}"
"""
}
Now, regardless of how long a process P2 takes to produce an output, process P3 will only produce results once both ChannelB and ChannelC have matching items (the first item) in them.
5.9.5. 6.5. Copying channels¶
You often need to copy a channel
process do {
...
output:
file ("x.*") into out_ch
...
}
out_ch.separate(a_ch, b_ch, c_ch)
Alternatively:
process do {
...
output:
file ("x.*") into (a_ch, b_ch, c_ch)
}
5.10. 7. Practical Execise: Using the H3ABioNet Variant Calling Pipeline¶
Exercise 7: We will now get to play with a Nextflow bioinformatic pipeline for variant calling. The workflow is available in this GitHub repository. Instructions on how to obtain and use the workflow are detailed on the GitHub repository.